Accurate measurement of AAV genome titer throughout the development and production for gene therapy is important to ensure the effectiveness of the final product. Conventionally, this task has been accomplished using qPCR and more recently digital PCR (ddPCR) methods. Each has its own benefits and downsides.
While qPCR offers a 4 logs dynamic range, it has several drawbacks – significant variability with CV (coefficients of variation) reaching up to 30%, susceptibility to inhibitory factors such as PCR inhibitors, adverse impact of extraneous DNA, lengthy analysis times and the necessity to establish a standard curve.
The ddPCR on the other hand presents several advantages viz. it eliminates the need for a standard curve, is accurate and provides high precision ranging from 3% to 20%. However, this impressive accuracy and precision is attainable within a limited quantitative dynamic range of approx. 2logs. The ddPCR’s multiplexing capability facilitates the examination of genome integrity, thus making it very useful. That said, the ddPCR process involves extended assay times, a relatively intricate multi-step workflow, and vulnerability to PCR limitations, posing challenges for routine application. Notably, 100-to-1,000,000-fold dilution is needed for samples to fall within the working range and to minimize matrix effects seen in in-process samples, introducing error and extended sample processing time. The cost per sample can also be substantial.
The above observations motivated us to develop a fast, easy, and accurate assay for in-process samples based on a novel method that excels in precision, speed, accuracy and most importantly simplicity.
Our groundbreaking approach revolves around DNA hybridization, immunochemistry and biolayer interferometry, encompassing a streamlined two-step procedure involving lysis and hybridization within a single tube, succeeded by detection using a biosensor.
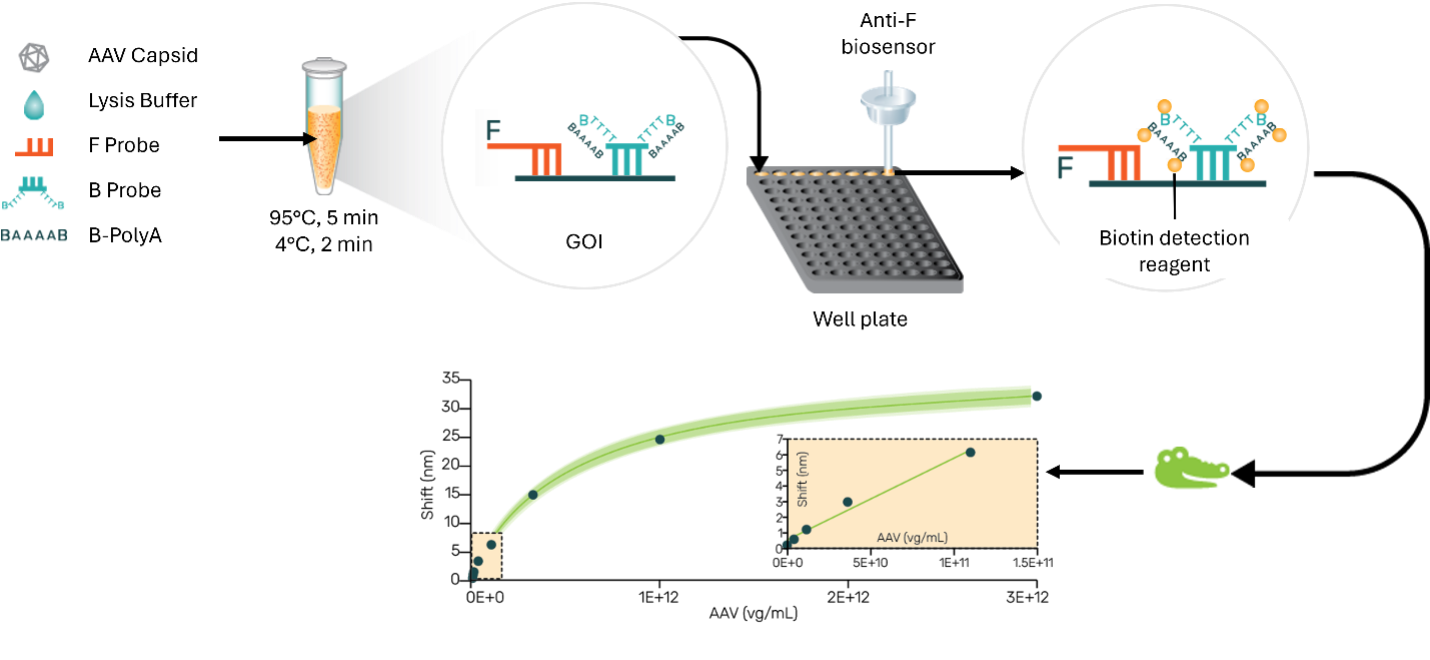
Central to the methodology are two oligo probes—an approximate 40 nt fluorescein-labelled probe and a 40 nt SuPlex probe. The SuPlex oligo includes a 30 Thymine oligonucleotide bearing biotin at both the 5′ and 3′ ends. The target region for these probes can be located within the Gene of Interest (GOI) or the promoter/enhancer regions. The lysis and hybridization steps involve heating at 950 C, promptly followed by rapid cooling to 50 C. The resultant hybridized sample is quantitated employing an anti-fluorescein biosensor.
The assay offers a quantitative dynamic range of 5E+9 to 5E+12 and delivers precision ranging from 3% to 9%. The assay time is mere 30 minutes. The inherent robustness of this method makes it compatible with DNase1, proteinase K, extraneous DNA, PCR inhibitors and in-process buffers, rendering it highly suitable for both research and manufacturing. Notably, another unique feature is the ability to quantify individually both positive and negative strands of the packed genome. Also, our preliminary data using oligo probes for GFP, CMV promoter, CMV enhancer and SV40 regions shows the potential for determining genome integrity.
So, while QPCR and digital PCR are currently the main tools for AAV genome titer assay, the DNA hybridization-biolayer interferometry approach described here shows significant benefits over the PCR methods. Its ability to characterize packed genome content deeper by providing strand specific titers, and robustness against various inhibitory factors positions it as an asset in both research and manufacturing contexts. Further, we have applied the same methodology for direct RNA titer without reverse transcription in Lentivirus.